The Harvard Gazette: New drug target for Rett syndrome
Researchers identify potential new route to therapy
Harvard Stem Cell Institute (HSCI) researchers have identified a faulty signaling pathway that, when corrected in mice, ameliorates the symptoms of Rett syndrome, a devastating neurological condition. The findings could lead to the discovery of compounds or drugs that may benefit children affected by the disease, says neurobiologist Jeffrey Macklis, a member of the HSCI Executive Committee.
The research was published recently in Nature Communications. Noriyuki Kishi and Jessica MacDonald, both recent postdoctoral fellows in the Macklis laboratory, are co-first authors. Macklis, who directed the work, is the Max and Anne Wien Professor of Life Sciences in the Department of Stem Cell and Regenerative Biology, and Center for Brain Science at Harvard University.
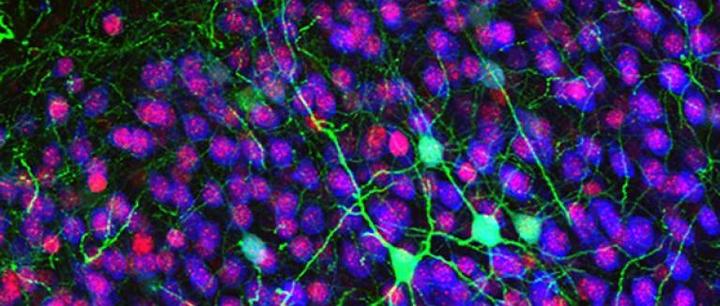
Callosal projection neurons (green) in the cerebral cortex. In 2004, Jeffrey Macklis’ lab was the first to describe abnormal development in this type of neuron.Photo courtesy of Jessica MacDonald and Jeffrey Macklis
Rett syndrome is a relatively common neurodevelopmental disorder, the second most common cause of intellectual disability in girls after Down syndrome; it is associated with a dysfunctional gene on the X chromosome. Boys with Rett syndrome are rare, because male fetuses who carry the mutations on their one X chromosome usually have prenatally lethal forms of the disease.
Girls with Rett syndrome appear to develop relatively normally for the first six to 18 months of life, but then regress; they tend to lose their ability to speak and the purposeful use of their hands, withdraw from social situations, and wring their hands.
Austrian physician Andreas Rett first described the disorder in 1966, but it wasn’t until 1999 that Huda Zoghbi and her lab at Baylor College of Medicine identified mutations in the gene MECP2 as the root cause of Rett syndrome. MECP2, however, turns a very large number of genes on and off throughout the entire body, so it has been a long-standing puzzle why children with Rett syndrome have this very specific and reproducible developmental cognitive brain disorder.
“My view was that MECP2 mutation in Rett syndrome disrupts so many genes and their protein products that we weren’t going to find a single gene that we could fix to help girls with Rett,” said Macklis, former program head of HSCI’s Nervous System Diseases Program, and an Allen Distinguished Investigator of the Paul G. Allen Family Foundation. “But if we found a disrupted, improperly regulated signaling pathway that was ‘drug-able,’ that affected enough of the girls’ pathology, we might be able to make them dramatically functionally better with already available therapeutics — and that might make a real difference in their lives and their families’ lives.”
Instead of concentrating on the MECP2 gene, Macklis’ group focused on neurons he knew were “abnormal and implicated in Rett syndrome and autism spectrum disorders,” and in 2004, his lab was the first to describe abnormal development in this type of neuron. These neurons, called inter-hemispheric callosal projection neurons (CPNs), have shorter, less-developed dendrites, or “receiving antennas,” in mice with the Rett gene mutations and in individuals with Rett syndrome.